
Script
📚 Catalogue
De-meta-analysis
It is also possible to exclude the effects of one cohort from GWAS summary statistics, using a de-meta-analysis:
\[\hat {\beta}_{DE-META} = \hat{\beta}_{META} - SE^2_{\hat{\beta}_{DE-META}} \sum_c \frac{\hat {\beta}_{c} - \hat{\beta}_{META} }{SE^2_{\hat{\beta}_{c}}}\] \[SE_{\hat{\beta}_{DE-META}} = \sqrt{\frac{1}{\frac{1}{SE^2_{\hat{\beta}_{META}}}-\sum_c \frac{1}{SE^2_{\hat{\beta}_{c}}}}}\]🧠 Let’s we implement using R script
rescale_b_se <- function(gwas, case, control){
names(gwas) = c("SNP","A1","A2","freq","b","se","P","N")
n_eff = as.integer(4*case*control/(case + control))
p = gwas$freq
z = gwas$b / gwas$se
se = 1/sqrt(2*p*(1-p)*(n_eff+z^2))
b = z*se
gwas$b = b
gwas$se = se
gwas$N = n_eff
return(gwas)
}
de_meta <- function(meta, sub_c){
if (!requireNamespace("logger", quietly=TRUE)){ install.packages("logger") }
lapply(c("dplyr","data.table","logger"), function(pkg) suppressWarnings(library(pkg,character.only=TRUE)))
log_info("Start to check the input meta...")
log_info("Start to check SNP effect size")
if(any(meta$beta_meta == 0)){
Delmeta = meta[meta$beta_meta == 0, ]
log_info(paste("The meta file contained", nrow(Delmeta), "SNPs that effect size = 0"))
meta = meta[!(SNP %in% Delmeta$SNP)]
log_info("We will delete these SNPs")
}else{ log_info("No SNPs effect size = 0") }
log_info("Start to check P value")
if(any(meta$p > 1 | meta$p < 0)){
Delmeta = meta[meta$p > 1 | meta$p < 0, ]
log_info(paste("The meta file contained", nrow(Delmeta), "SNPs that Pvalue > 1 or < 0"))
meta = meta[!(SNP %in% Delmeta$SNP)]
log_info("We will delete these SNPs")
}else{ log_info("No SNPs Pvalue > 1 or < 0") }
log_info("Start to check the input sub_c...")
log_info("Start to check SNP effect size")
if(any(sub_c$beta_c == 0)){
Delmeta = sub_c[sub_c$beta_c == 0, ]
log_info(paste("The sub_c file contained", nrow(Delmeta), "SNPs that effect size = 0"))
sub_c = sub_c[!(SNP %in% Delmeta$SNP)]
log_info("We will delete these SNPs")
}else{ log_info("No SNPs effect size = 0") }
log_info("Start to merge the meta and sub_c by SNP")
combined_data <- merge(meta,sub_c,by="SNP")
log_info("Start to keep meta only SNPs")
meta_only <- meta[meta$SNP %in% setdiff(meta$SNP,sub_c$SNP), ]
meta_only [, n_meta := n_meta - unique(sub_c$N)]
setDT(meta_only)
setnames(meta_only, c("SNP","A1","A2","freq","b","se","p","N"))
log_info(paste("There were", nrow(meta_only), "SNPs only occured in meta"))
log_info("Start to fix the effect allele in meta and sub_c")
combined_data[, beta_c := ifelse((A1_m==A1_c & A2_m==A2_c),beta,ifelse(A1_m==A2_c & A2_m==A1_c,-beta,NA_real_))]
if(any(is.na(combined_data$beta_c))) {
Deldt = subset(combined_data, is.na(combined_data$beta_c))
log_info(paste("There were", nrow(Deldt), "SNPs have unmatched alleles, will delete these SNPs!"))
log_info(paste("In these SNPs, the lowest P value is", min(Deldt$p)))
combined_data = combined_data[!(SNP %in% Deldt$SNP)]
}
log_info("Start to demeta SNP effect size and se")
se_meta_mean <- mean(combined_data$se_meta, na.rm=TRUE)
combined_data[, se_demeta := fifelse((1/se_meta^2 - 1/se_c^2) > 0, sqrt(1/(1/se_meta^2 - 1/se_c^2)),se_meta_mean)]
combined_data[, beta_demeta := beta_meta-se_demeta^2 * (beta_c-beta_meta)/se_c^2]
if (any(combined_data$beta_demeta == 0)) {
log_info("There may be generate zero beta after demeta!")
Deldt = combined_data[combined_data$beta_demeta == 0, ]
log_info(paste("There were", nrow(Deldt), "SNPs has been deleted casued by the demeta value is zero!"))
combined_data = combined_data[!(SNP %in% Deldt$SNP), ]
}
log_info("Start to create effect N of demeta")
combined_data[, N_eff := n_meta - N]
common_SNP <- combined_data[, c("SNP","A1_m","A2_m","freq","beta_demeta","se_demeta","p","N_eff")]
setDT(common_SNP)
setnames(common_SNP, c("SNP","A1","A2","freq","b","se","p","N"))
log_info("Start to combine meta only SNPs and demeta SNPs to one")
outfile <- rbind(common_SNP,meta_only)
log_info("Start to calculate P value based on beta and se")
outfile$p <- 2 * (1 - pnorm(abs(outfile$b / outfile$se)))
log_info("Demeta analysis is completed!")
return(outfile)
}
🧪 Just use it as follow: in this case, we using hapmap 3 SNP to do de-meta
library(data.table)
hm3 = fread("listHM3.txt", col.names="SNP")
meta = fread("ARHL_MVP_AJHG_BBJ_reformatMETAL.txt")
meta_hm3 = merge(meta, hm3, by="SNP")
names(meta_hm3) = c("SNP","A1_m","A2_m","freq","beta_meta","se_meta","p","n_meta")
## rescale beta se for sub cohort data
sub_c = fread("2247_1.v1.1.fastGWA", select=c("SNP","A1","A2","AF1","BETA","SE","P","N"))
sub_c_hm3 = merge(sub_c, hm3, by="SNP")
re_sub_c_hm3 = rescale_b_se(sub_c_hm3, 114318, 323449)
names(re_sub_c_hm3) = c("SNP","A1_c","A2_c","freq_c","beta","se_c","p_c","N")
# run demeta for meta and ajhg
demeta_meta_hm3 = de_meta(meta_hm3, re_sub_c_hm3)
out_meta_demeta_hm3 = demeta_meta_hm3[which(demeta_meta_hm3$N > 100000),]
fwrite(out_meta_demeta_hm3, file="Demeta_raw_hm3.txt", sep="\t")
SMR plot for multi tissue in one fig
🧠 When you want draw multi tissue SMR result in one fig, this script will meet your require
#***********************************************#
# File : CreateMultiTissueSMRPlotData.r #
# Time : 14Apr2025 18:00:51 #
# Author : Loren Shi #
# Mails : crazzy_rabbit@163.com #
# link : https://github.com/Crazzy-Rabbit #
#***********************************************#
ExtractProbe <- function(QTL, chr, centerbp, window=10e5, probes, tissue){
# 1.generate probe list in range
start = centerbp - window;
end = centerbp + window;
QTLepi = read.delim(paste0(QTL,".epi"),head=F)
QTLindex = which(QTLepi$V1==chr & QTLepi$V4>=start & QTLepi$V4<=end)
QTLout = paste0(dir, "/", basename(QTL), "_", probes) # probe list filename
write.table(QTLepi[QTLindex,2], QTLout,col.names=FALSE,row.names=FALSE,sep="\t",quote=FALSE)
# 2.extract besd files
SMR="~/software/smr-1.3.1/smr"
system(paste(SMR, "--beqtl-summary", QTL,
"--chr", chr,
"--extract-probe", QTLout,
"--thread-num 10",
"--make-besd",
"--out", QTLout))
# 3.update the probe names in epi file, probe as tissue: ENSG00000122786
epi = read.table(paste0(QTLout, ".epi"), head=FALSE)
epi$V2 = paste0(tissue, "-", epi$V2)
write.table(epi, paste0(QTLout, ".epi"), col.names=FALSE,row.names=FALSE,sep="\t",quote=FALSE)
return(QTLout)
}
CreateMultiTissueSMRPlotDt <- function(esi_profix_list, tissue, probes, ref, gwas, windows=2000, outprefix){
library(data.table)
# the loop to match esi file pos
esi_list = lapply(esi_profix_list, function(prefix) {
fread(paste0(prefix, ".esi"))
})
n=length(esi_list)
esiSet = unique(do.call(rbind, esi_list))
snpSet = unique(unlist(lapply(esi_list, function(x) x$V2)))
idx = match(snpSet, esiSet$V2)
refSet = data.frame(snpSet, A1=esiSet$V5[idx], A2=esiSet$V6[idx])
idx_list = lapply(esi_list, function(x) match(x$V2, esiSet$V2))
for (i in seq_along(esi_list)) {
idxs = idx_list[[i]]
esi_list[[i]]$V5 = refSet$A1[idxs]
esi_list[[i]]$V6 = refSet$A2[idxs]
outfile = paste0(esi_profix_list[i], ".esi")
write.table(esi_list[[i]],outfile,col.names=FALSE,row.names=FALSE,sep="\t",quote=FALSE)
}
# merge multi tissue besd file
SMR="~/software/smr-1.3.1/smr"
genelist="~/script/plot_smr/glist_hg19_refseq.txt"
flist = paste0(outprefix, ".list")
writeLines(esi_profix_list, flist)
system(paste(SMR, "--besd-flist", flist,
"--make-besd",
"--out", outprefix))
# extract plot dt
new_probe = paste0(tissue, "-", probes)
system(paste(SMR, "--beqtl-summary", outprefix,
"--gwas-summary", gwas,
"--bfile", ref,
"--plot",
"--diff-freq-prop 0.2",
"--probe", new_probe,
"--probe-wind", windows,
"--gene-list", genelist,
"--out", outprefix))
}
please replace the SMR dir when you use this script ! ! !
🧪 Example for using:
# index for below used
reference="~/Wulab_share/1000GenomePhase3_Ref_hg37/g1000_eas/g1000_eas"
Adipose_Subcutaneous="/~/Wulab_share/SMR_xQTL_besd/eqtl_all_tissue_GTEx/Adipose_Subcutaneous"
Skin_Sun_Exposed_Lower_leg="~/Wulab_share/SMR_xQTL_besd/eqtl_all_tissue_GTEx/Skin_Sun_Exposed_Lower_leg"
gwas='~/Wulab_project/NSOC/NSOC_filter_maf.txt'
dir="~/Wulab_project/NSOC/CALD1_eQTL"
source("~/script/plot_smr/CreateMultiTissueSMRPlotData.r")
# extract target probe and update the probe name in new epi file
file_idx1 = ExtractProbe(QTL=Adipose_Subcutaneous, chr=7, centerbp=134.429e6, window=10e5,
probes="ENSG00000122786", tissue="Adipose_Subcutaneous")
file_idx2 = ExtractProbe(QTL=Skin_Sun_Exposed_Lower_leg, chr=7, centerbp=134.429e6, window=10e5,
probes="ENSG00000122786", tissue="Skin_Sun_Exposed_Lower_leg")
# match pos in each esi file and merge these besd file, then generate multi tissue SMR plot file
esi_profix_list=c(file_idx1, file_idx2)
CreateMultiTissueSMRPlotDt(esi_profix_list=esi_profix_list, tissue="Adipose_Subcutaneous", probes="ENSG00000122786",
ref=reference, gwas=gwas, windows=2000, outprefix="multiTissue_eQTL_CALD1")
#====================================#
# plot
#====================================#
source("E:/Shi/ALL_of_my_Job/WCH_script/SMRplot/plot_epiSMR_LorenShi.r")
SMRData = ReadSMRData("multiTissue_eQTL_CALD1.Adipose_Subcutaneous-ENSG00000122786.txt")
pdf('CALD1_multi_tissue.ENSG00000122786.pdf',width = 10,height = 15)
SMRLocusPlot(data=SMRData, smr_thresh=2.44e-4, heidi_thresh=0.01, plotWindow=400, max_anno_probe=4,
probeNEARBY=c("Skin_Sun_Exposed_Lower_leg-ENSG00000122786"), highlight=c("rs17168118","rs10488465", "rs4732060"),
anno_self=T, annoSig_only=F, epi_plot=TRUE, funcAnnoFile = "E:/Shi/plot_smr/funcAnno.RData")
dev.off()
Manhattan plot for GWAS or others
🧠 When you want draw Manhattan plot, follow scripts will meet your require
1 gwaslab
More detail please see gwaslab
import gwaslab as gl
import pandas as pd
import matplotlib.pyplot as plt
# 1. data read
gwas = gl.Sumstats("ARHL_MVP_AJHG_BBJ_reformatMETAL_addchr.gz",
snpid="SNP",
chrom="CHR",
pos="POS",
ea="A1",
nea="A2",
eaf="freq",
beta="beta",
se="SE",
p="p",
n="N",
build="19")
# 2. plot manhattan with annota
df = pd.read_csv("novel_snp_ARHL.txt", sep="\t")
anno_list = df["SNP"].tolist()
gwas.plot_mqq(mode="m", skip=0, sig_line_color="red", fontsize=12, marker_size=(5,5),
anno="GENENAME", anno_style="expand", anno_set=anno_list, anno_fontsize=12, repel_force=0.01, arm_scale=1,
xtight=True, ylim=(0,38), chrpad=0.01, xpad=0.05, # cut=40, cut_line_color="white",
fig_args={"figsize": (18, 5), "dpi": 500},
save="mqq_plot.png", save_args={"dpi": 500}, check=False, verbose=False)
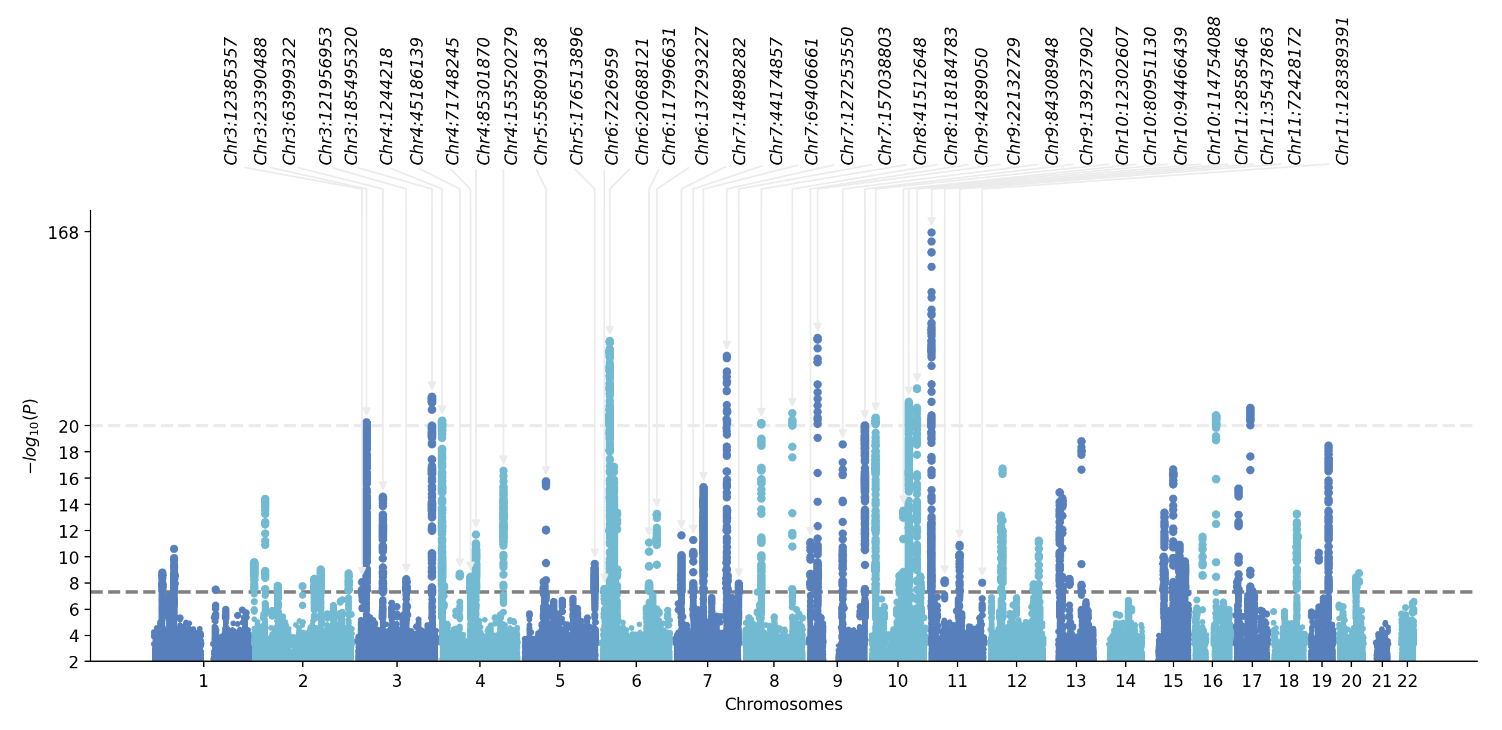
2 CMplot
More detail please see CMplot
# file columns should has SNP CHR BP P
args <- commandArgs(TRUE)
ptype <- args[1]
infile <- args[2]
outname <- args[3]
library(CMplot)
library(data.table)
library(dplyr)
data <- fread(infile) %>% filter(complete.cases(.))
if (!all(c("SNP", "CHR", "BP", "P") %in% names(data))) stop("The file you provide not contains columns SNP, CHR, BP, and P")
pltdt <- subset(data, select=c(SNP, CHR, BP, P))
plt_filter <- na.omit(pltdt[pltdt$P > 0 & pltdt$P < 1, ]) # remove na and keep 0 < p < 1
my_color <- c('#1B2C62', '#4695BC')
manhattan <- function (plt, out, ylim=NULL, dpi=500, height=5, width=15, highlight=NULL, highlight.pch=19) {
CMplot(plt, type="p", plot.type="m", LOG10=TRUE, ylim=ylim,
file="jpg", file.name=out, file.output=TRUE, dpi=dpi,
threshold=5e-8, threshold.col="red", threshold.lwd=2, threshold.lty=2,
highlight=highlight, highlight.pch=highlight.pch,
col=my_color, band=0,
cex=0.6, signal.cex=0.6, height=height, width=width)
}
qqplot <- function(plt, out, dpi=500, width=10, height=10){
CMplot(plt, plot.type="q", LOG10=TRUE, file="jpg", file.name=outname, dpi=dpi,
file.name=out, verbose=TRUE, cex=0.8, signal.cex=0.8, width=width, height=height,
threshold.col="red", threshold.lty=2, conf.int=TRUE, conf.int.col=NULL, file.output=TRUE)
}
if (ptype == "manhattan") {
if (any(-log10(plt_filter$P) > 50)){
manhattan(plt=plt_filter, out=outname, ylim=c(0, 50))
} else {
manhattan(plt=plt_filter, out=outname)
}
} elseif (ptype == "qqplot") {
qqplot(plt=plt_filter, out=outname)
}
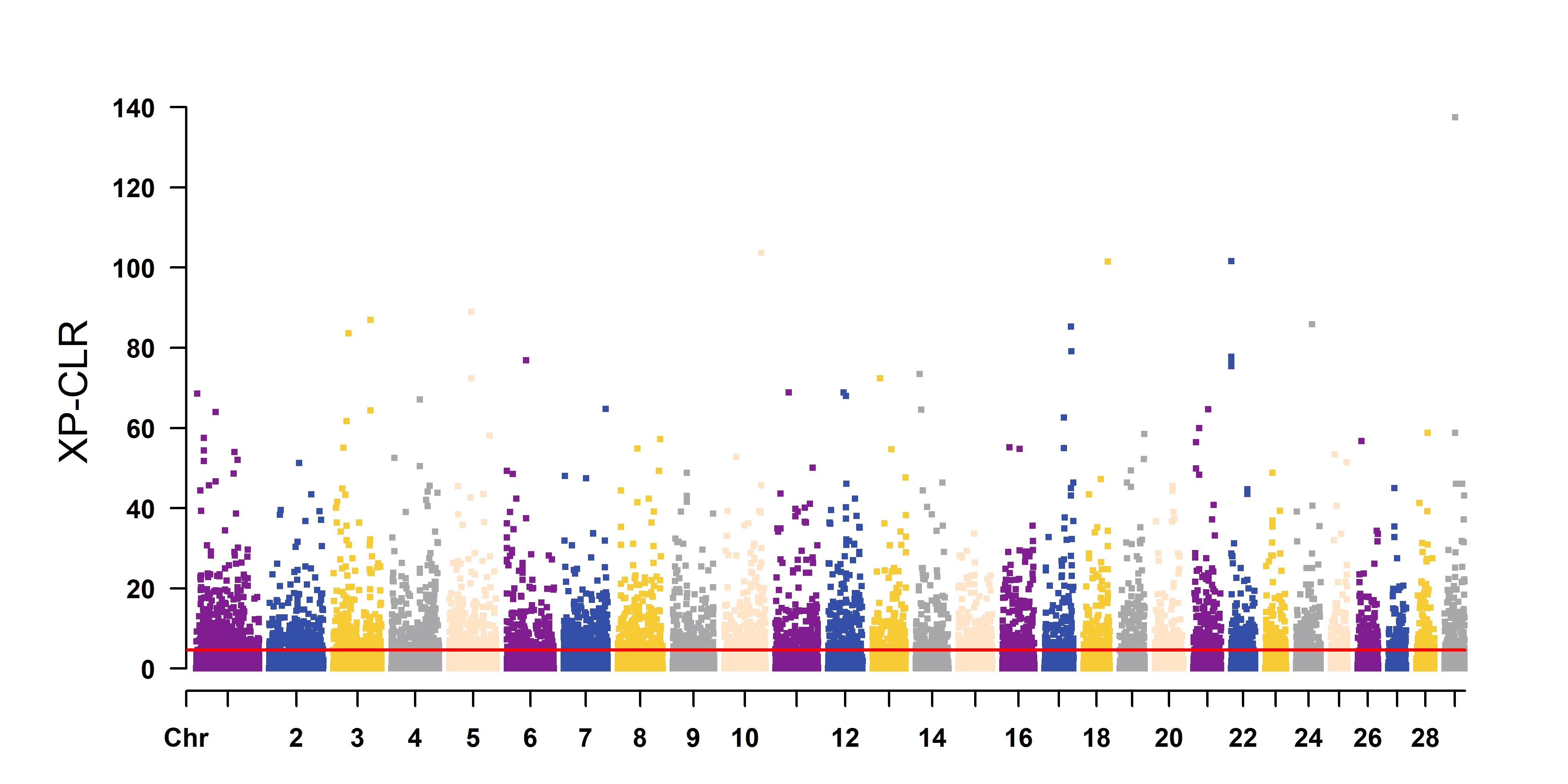
3 ggplot2
library(data.table)
library(ggplot2)
library(dplyr)
dt = fread(infile)[, c("CHR", "BP", "P")]
chr_lengths <- dt %>%
group_by(CHR) %>%
summarise(chr_len = max(BP))
data <- dt %>%
arrange(CHR, BP) %>%
mutate(chr_cumsum = cumsum(c(0, head(chr_lengths$chr_len, -1)))[CHR]) %>%
mutate(BPcum = BP + chr_cumsum)
axis_set <- data %>%
group_by(CHR) %>%
summarise(center = (max(BPcum) + min(BPcum)) / 2)
p = ggplot(data, aes(x=BPcum, y=-log10(P), color=factor(CHR))) +
geom_point(alpha=0.9) +
geom_hline(yintercept=-log10(5e-8), color="red", linetype="dashed") +
scale_color_manual(values=rep(c("#1B2C62", "#4695BC"), 22)) +
scale_x_continuous(label=axis_set$CHR,
breaks=axis_set$center,
expand=c(0, 0)) +
scale_y_continuous(expand=c(0, 0)) +
labs(x="Chromosome", y=expression(-log[10](P))) +
theme_classic() +
theme(legend.position="none",
axis.text.x = element_text(face="bold", size=20, color="black"),
axis.text.y = element_text(face="bold", size=20, color="black"),
axis.title.x = element_text(face="bold", size=20, margin=margin(t=10), color="black"),
axis.title.y = element_text(face="bold", size=20, margin=margin(t=10), color="black"),
axis.line = element_line(color="black"),
axis.ticks = element_line(color="black"),
axis.ticks.length = unit(0.3, "cm"))
ggsave("my.png", p, height = 8, width = 23, dpi=300)
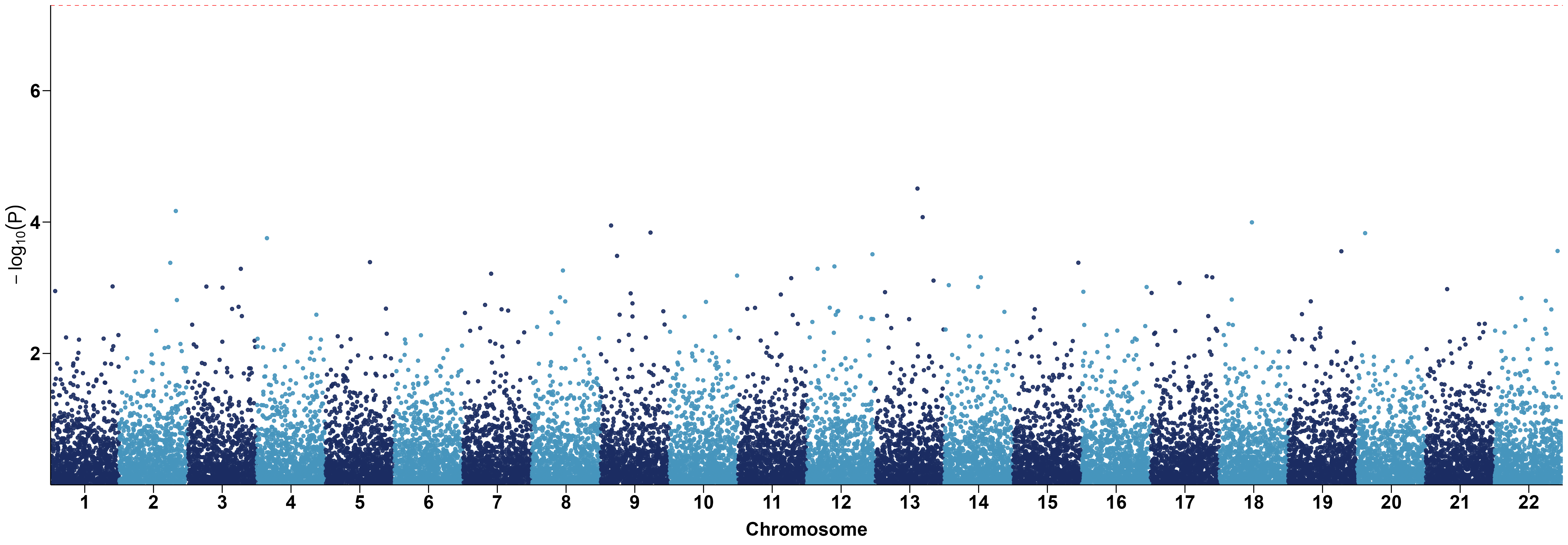
4 Python
# -*- coding: utf-8 -*-
"""
Created on Mon Jan 15 20:28:05 2018
@author: Caiyd
"""
import json
import click
CONTEXT_SETTINGS = dict(help_option_names=['-h', '--help'])
import numpy as np
import pandas as pd
import matplotlib
matplotlib.use('Agg')
import matplotlib.pyplot as plt
import seaborn as sns
from itertools import cycle
def load_allchrom_data(infile, chr_col, loc_col, val_col, log_trans, neg_logtrans):
"""
infile contain several chromosomes
"""
df = pd.read_csv(infile,
sep='\t',
usecols = [chr_col, loc_col, val_col],
dtype={chr_col: str,
loc_col: float,
val_col: float})
if log_trans:
df.loc[:, val_col] = np.log10(df[val_col].values)
elif neg_logtrans:
df.loc[:, val_col] = -np.log10(df[val_col].values)
df.dropna(inplace=True)
return df
def load_cutoff(cutoff, log_trans, neg_logtrans):
with open(cutoff) as f:
cutoff = {}
for line in f:
tline = line.strip().split()
value = float(tline[1])
if log_trans:
value = np.log10(value)
elif neg_logtrans:
value = -np.log10(value)
cutoff[tline[0]] = value
return cutoff
def plot(df, chr_col, loc_col, val_col, xlabel, ylabel, ylim, invert_yaxis, top_xaxis, cutoff,
highlight, outfile, ticklabelsize, figsize, axlabelsize, markersize, fill_regions,
windowsize):
sns.set_style('white', {'ytick.major.size': 3, 'xtick.major.size': 3})
fig, ax = plt.subplots(1, 1, figsize=figsize)
# offsets = {}
loc_offset = 0
xticks = []
xticklabels = []
# for fill_between
valmax = df[val_col].max()
valmin = df[val_col].min()
if ylim:
y1, y2 = ylim
else:
y1 = valmin
y2 = valmax * 1.05
# plot val
for chrom, color in zip(df[chr_col].unique(), cycle(['#1B2C62', '#4695BC'])):
# offsets[chrom] = loc_offset
tmpdf = df.loc[df[chr_col]==chrom, [loc_col, val_col]]
tmpdf[loc_col] += loc_offset
xticklabels.append(chrom)
xticks.append(tmpdf[loc_col].median())
tmpdf.plot(kind='scatter', x=loc_col, y=val_col, ax=ax, s=markersize, color=color, marker='o')
if isinstance(highlight, pd.DataFrame):
hdf = highlight.loc[highlight[chr_col]==chrom, [loc_col, val_col]]
if hdf.shape[0] > 0:
hdf[loc_col] += loc_offset
hdf.plot(kind='scatter', x=loc_col, y=val_col, ax=ax, s=markersize, color='#55A868', marker='o')
if cutoff:
ax.hlines(cutoff[chrom], tmpdf[loc_col].values[0], tmpdf[loc_col].values[-1], colors='#CEAF7D', linestyles='dashed')
# fill_between region
if isinstance(fill_regions, pd.DataFrame):
regions = fill_regions.loc[fill_regions['chrom']==chrom, ['start', 'end']].values
if regions.shape[0] > 0:
regions += loc_offset
for region in regions:
# region: list [float, float]
ax.fill_between(region, y1, y2, color='grey', alpha=0.6)
# plot mean value line
if windowsize:
tmpdf['window_index'] = tmpdf[loc_col] // windowsize
tmpdf.groupby('window_index').agg({loc_col: np.median,
val_col: np.median}).plot(x=loc_col, y=val_col, kind='line', ax=ax)
loc_offset = tmpdf[loc_col].values[-1] # assume loc is sorted
ax.set_xlabel(xlabel, fontsize=axlabelsize)
ax.set_ylabel(ylabel, fontsize=axlabelsize)
ax.xaxis.set_ticks_position('bottom')
ax.yaxis.set_ticks_position('left')
plt.xticks(xticks, xticklabels)
ax.spines['left'].set_linewidth(2)
ax.spines['bottom'].set_linewidth(2)
plt.subplots_adjust(left=0.05, right=0.99)
plt.tick_params(axis='both', which='both', direction='out', width=2, length=6, labelsize=ticklabelsize)
ax.set_xlim([df[loc_col].values[0], tmpdf[loc_col].values[-1]])
# adjust ylim
if ylim:
ax.set_ylim(ylim)
# adjust axis
if invert_yaxis:
ax.invert_yaxis()
if top_xaxis:
ax.xaxis.tick_top()
ax.spines['right'].set_visible(False)
ax.spines['bottom'].set_visible(False)
ax.xaxis.set_label_position('top')
else:
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
# adjust font size
for label in (ax.get_xticklabels() + ax.get_yticklabels()):
label.set_fontsize(ticklabelsize)
# hide legend
ax.legend().set_visible(False)
plt.savefig(f'{outfile}', dpi=450)
plt.close()
@click.command(context_settings=CONTEXT_SETTINGS)
@click.option('--infile', help='tsv文件,包含header')
@click.option('--chr-col', help='染色体列名')
@click.option('--loc-col', help='x轴值列名')
@click.option('--val-col', help='y轴值列名')
@click.option('--log-trans', is_flag=True, default=False, help='对val列的值取以10为底的对数')
@click.option('--neg-logtrans', is_flag=True, default=False, help='对val列的值取以10为底的负对数(画p值)')
@click.option('--outfile', help='输出文件,根据拓展名判断输出格式')
@click.option('--xlabel', help='输入散点图x轴标签的名称')
@click.option('--ylabel', help='输入散点图y轴标签的名称')
@click.option('--ylim', nargs=2, type=float, default=None, help='y轴的显示范围,如0 1, 默认不限定')
@click.option('--invert-yaxis', is_flag=True, default=False, help='flag, 翻转y轴, 默认不翻转')
@click.option('--top-xaxis', is_flag=True, default=False, help='flag, 把x轴置于顶部, 默认在底部')
@click.option('--cutoff', default=None, help='两列,第一列染色体号,第二列对应的阈值')
@click.option('--highlight', default=None, help='和infile相同格式的文件,在该文件中的点会在曼哈顿图中单独高亮出来,default=None')
@click.option('--ticklabelsize', help='刻度文字大小', default=10)
@click.option('--figsize', nargs=2, type=float, help='图像长宽, 默认15 5', default=(15, 5))
@click.option('--axlabelsize', help='x轴y轴label文字大小', default=10)
@click.option('--markersize', default=6, help='散点大小, default is 6', type=float)
@click.option('--chroms', default=None, help='只用这些染色体,e.g --chr 6 --chr X will only plot chr6 and chrX.', multiple=True)
@click.option('--fill-regions', default=None, help='fill between regions in this <bed file> (no header)')
@click.option('--windowsize', default=None, help='draw mean value in a specific window size', type=int)
def main(infile, chr_col, loc_col, val_col, log_trans, neg_logtrans, outfile,
xlabel, ylabel, ylim, invert_yaxis, top_xaxis, cutoff, highlight, ticklabelsize,
figsize, axlabelsize, markersize, chroms, fill_regions, windowsize):
"""
\b
曼哈顿图
使用infile中的chr_col和loc_col列作为x轴, 对应的val_col值画在y轴
输出outfile, 根据outfile指定的拓展名输出相应的格式
别人的脚本,我稍微改了下,出图更好看
"""
print(__doc__)
print(main.__doc__)
df = load_allchrom_data(infile, chr_col, loc_col, val_col, log_trans, neg_logtrans)
if chroms:
print('use chroms:\n%s' % '\n'.join(chroms))
chroms = set(chroms)
df = df.loc[df[chr_col].isin(chroms), :]
if highlight:
highlight = load_allchrom_data(highlight, chr_col, loc_col, val_col, log_trans, neg_logtrans)
if fill_regions:
fill_regions = pd.read_csv(fill_regions, sep='\t', header=None, names=['chrom', 'start', 'end'],
dtype={'chrom': str, 'start': float, 'end': float})
if cutoff:
cutoff = load_cutoff(cutoff, log_trans, neg_logtrans)
plot(df, chr_col, loc_col, val_col, xlabel, ylabel, ylim, invert_yaxis, top_xaxis, cutoff,
highlight, outfile, ticklabelsize, figsize, axlabelsize, markersize, fill_regions, windowsize)
if __name__ == '__main__':
main()
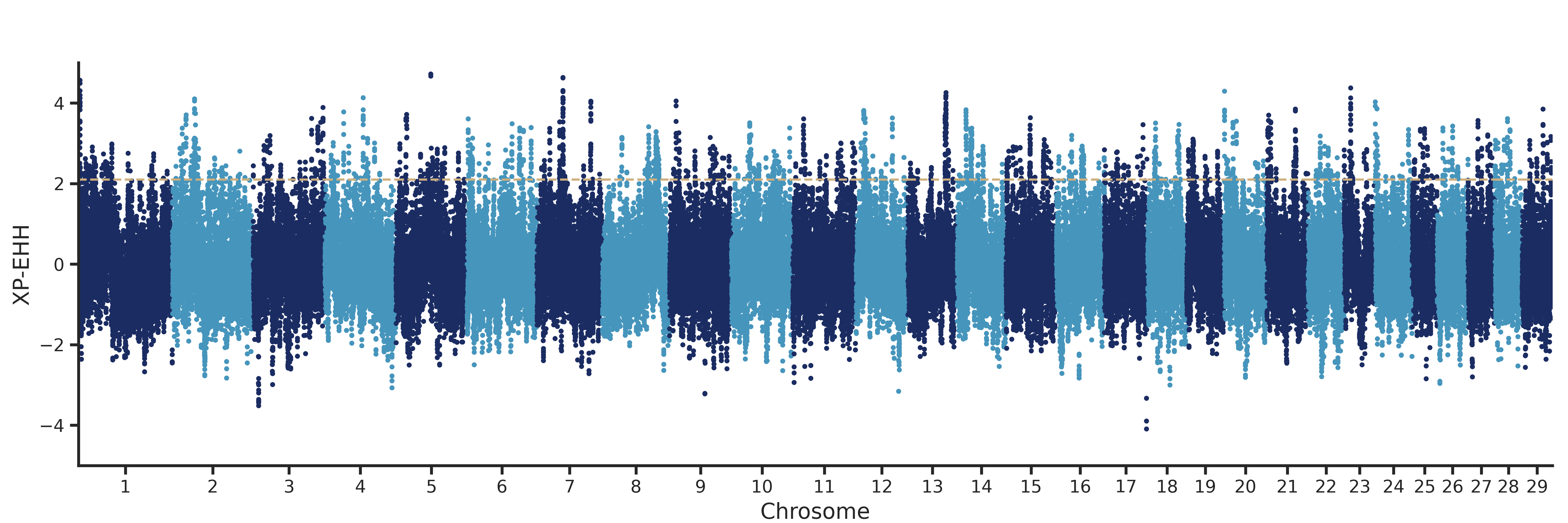
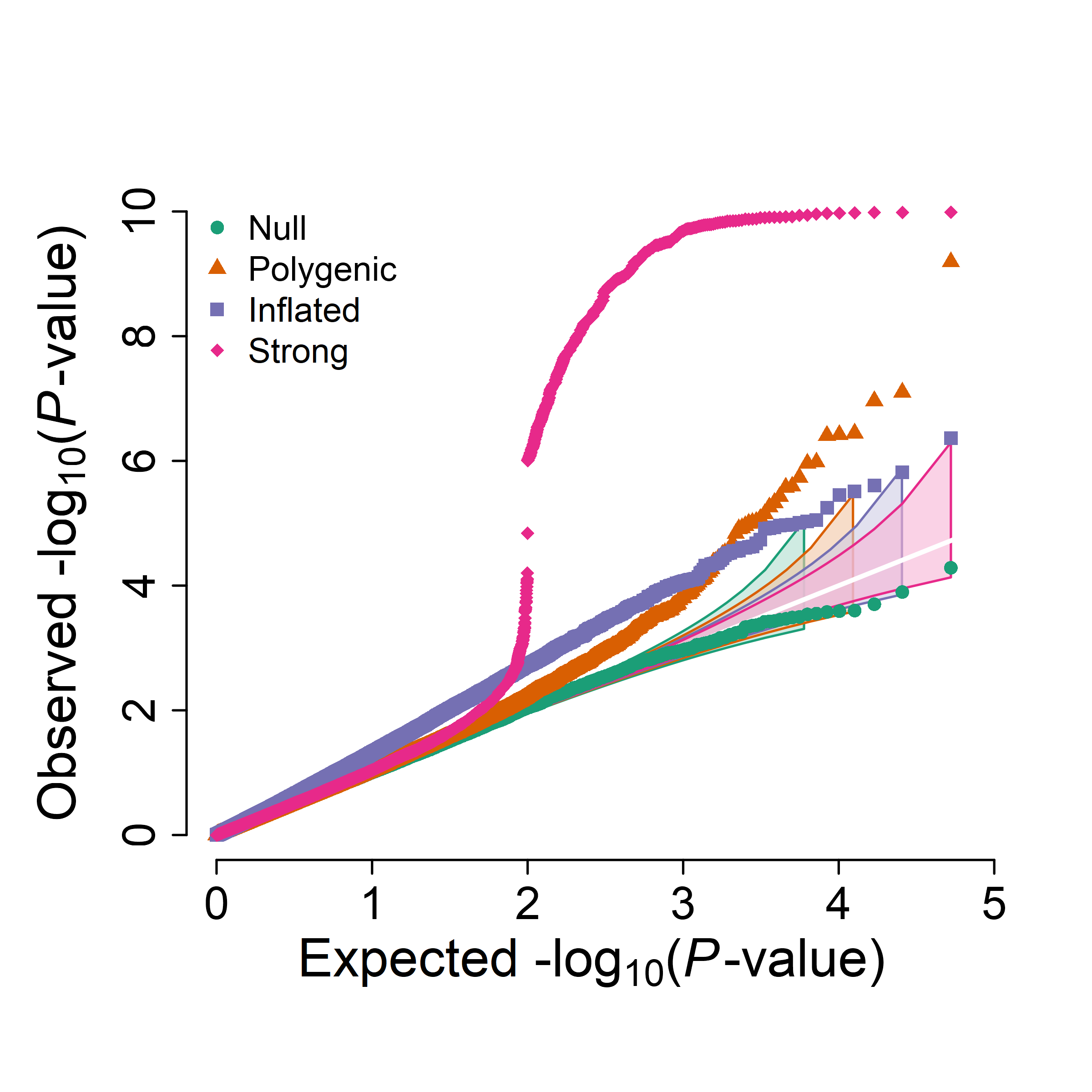
QQplot (single or multi trait)
1 single
qqplot = function(pval, ylim=0, lab=1.4, axis=1.2){
par(mgp=c(5,1,0))
p1 <- pval
p2 <- sort(p1)
n <- length(p2)
k <- c(1:n)
alpha <- 0.05
lower <- qbeta(alpha/2, k, n+1-k)
upper <- qbeta((1-alpha/2), k, n+1-k)
expect <- (k-0.05)/n
biggest <- ceiling(max(-log10(p2), -log10(expect)))
xlim <- max (-log10(expect)+0.1);
if (ylim==0) ylim=biggest;
plot(-log10(expect), -log10(p2), xlim=c(0, xlim), ylim=c(0, ylim),
ylab=expression(paste("Observed ", "-", log[10], "(", italic(P), "-value)", sep="")),
xlab=expression(paste("Expected ", "-", log[10], "(", italic(P), "-value)", sep="")),
type="n", mgp=c(2,0.5,0), tcl=-0.3, bty="n", cex.lab=lab, cex.axis=axis)
polygon(c(-log10(expect), rev(-log10(expect))), c(-log10(upper), rev(-log10(lower))),
col=adjustcolor("grey", alpha.f=0.5), border=NA)
abline(0,1,col="white", lwd=2)
points(-log10(expect), -log10(p2), pch=20, cex=0.6, col=2)
}
2 multi trait
qqplot_multi <- function(pval_list, ylim=0, lab=1.4, axis=1.2, p_floor=1e-300, cols=NULL, pch=20, cex=0.6,
legend=TRUE, legend_pos="topleft", legend_cex=0.9, show_ci=TRUE, ci_scale=1, ci_alpha=0.6, ci_border=TRUE, diag_col="white", ...) {
if (!is.list(pval_list)) pval_list=as.list(as.data.frame(pval_list))
trait = names(pval_list)
if(is.null(trait) || any(trait == "")){
trait=paste0("Trait", seq_along(pval_list))
names(pval_list)=trait
}
m = length(pval_list)
if(m < 1) stop("pval_list must contain at least one trait ! ! !")
recycle1m = function(x, nm, name){
if (length(x) == 1) return(rep(x, nm))
if (length(x) == nm) return(x)
stop(name, " length must be 1 or ", nm, ".")
}
ci_scale = recycle1m(ci_scale , m, "ci_xfrac")
ci_scale[ci_scale <= 0] = 0.01
ci_scale[ci_scale > 1] = 1
if(is.null(cols)){
if ("hcl.colors" %in% getNamespaceExports("grDevices")){
cols = grDevices::hcl.colors(m, palette="Dark 3")
} else {
cols = grDevices::rainbow(m)
}
}
cols = recycle1m(cols, m, "cols")
pch = recycle1m(pch, m, "pch")
lighten_col = function(col, amount=0.65, alpha=ci_alpha) {
rgb = grDevices::col2rgb(col) / 255
rgb2 = rgb + (1-rgb) * amount
grDevices::rgb(rgb2[1,], rgb2[2,], rgb2[3,], alpha=alpha)
}
dat=vector("list", m)
for(i in seq_len(m)){
p = pval_list[[i]]
p = p[is.finite(p) & !is.na(p)]
p = p[p <= 1 & p >= 0]
if (length(p) == 0) stop("Trait '", trait[i], "' has no valid P in [0,1].")
p[p == 0] = p_floor
p2 = sort(p)
n = length(p2)
k = seq_len(n)
alpha = 0.05
lower = qbeta(alpha/2, k, n+1-k)
upper = qbeta(1 - alpha/2, k, n+1-k)
expect = (k-0.05) / n
expect[expect <= 0] = min(expect[expect > 0])
dat[[i]] = list(x=-log10(expect), y=-log10(p2), y_low=-log10(upper), y_high=-log10(lower), n=n)
}
xlim = max(vapply(dat, function(d) max(d$x, na.rm=TRUE), numeric(1))) + 0.1
ymax = max(vapply(dat, function(d) max(d$y, d$y_high, na.rm=TRUE), numeric(1)))
if (ylim==0) ylim=ceiling(ymax)
par(mgp=c(5,1,0))
plot(NA, xlim=c(0, xlim), ylim=c(0, ylim),
ylab=expression(paste("Observed", " -", log[10], "(", italic(P), "-value", ")", sep="")),
xlab=expression(paste("Expected", " -", log[10], "(", italic(P), "-value", ")", sep="")),
type="n", mgp=c(2,0.5,0), tcl=-0.3, bty="n", cex.lab=lab, cex.axis=axis, ...)
if (show_ci){
for(i in seq_len(m)){
d = dat[[i]]
o = order(d$x)
x = d$x[o]
ylow = d$y_low[o]
yhigh = d$y_high[o]
x_ci = x * ci_scale[i]
ylow = ylow * ci_scale[i]
yhigh = yhigh * ci_scale[i]
ci_col = lighten_col(cols[i])
if (ci_border) {
border=cols[i]
} else {
border=NA
}
polygon(c(x_ci, rev(x_ci)), c(ylow, rev(yhigh)), col=ci_col, border=border)
}
}
abline(0, 1, col=diag_col, lwd=2)
for(i in seq_len(m)){
d = dat[[i]]
points(d$x, d$y, pch=pch[i], cex=cex, col=cols[i])
}
if (legend){
legend(legend_pos, legend=trait, col=cols, pch=pch, pt.cex=cex, bty="n", cex=legend_cex)
}
invisible(dat)
}
example
set.seed(20260108)
N <- 50000
# 1) 纯零假设(均匀分布)
p_null <- runif(N)
# 2) “多基因/少量真实信号”:97% null + 3% 偏小p
p_poly <- runif(N)
sig_poly <- rbinom(N, 1, 0.03) == 1
p_poly[sig_poly] <- rbeta(sum(sig_poly), shape1 = 0.4, shape2 = 1) # shape1<1 会产生更多小p
# 3) “膨胀”:|Z|整体偏大(sd>1),p会更偏小但不一定有真实峰
z_inf <- rnorm(N, mean = 0, sd = 1.2)
p_infl <- 2 * pnorm(-abs(z_inf))
# 4) “强信号”:1% 极强关联(生成极小p),并故意加入几个0
p_strong <- runif(N)
idx_strong <- sample.int(N, size = round(0.01 * N))
p_strong[idx_strong] <- 10^(-runif(length(idx_strong), min = 6, max = 10)) # 1e-6 到 1e-30
# p_strong[sample.int(N, 5)] <- 0 # 故意放几个0,测试防 Inf
pvals_list <- list(
Null = p_null,
Polygenic = p_poly,
Inflated = p_infl,
Strong = p_strong
)
png("qqplot_multi.png", width=2400, height=2400, res=500, type="cairo")
qqplot_multi(
pvals_list,
cols = c("#1b9e77", "#d95f02", "#7570b3", "#e7298a"),
pch = c(16, 17, 15, 18),
cex = c(0.8, 0.8, 0.8, 0.8),
ci_scale = seq(0.8, 1.0, length.out = 4),
ci_alpha=0.6)
dev.off()
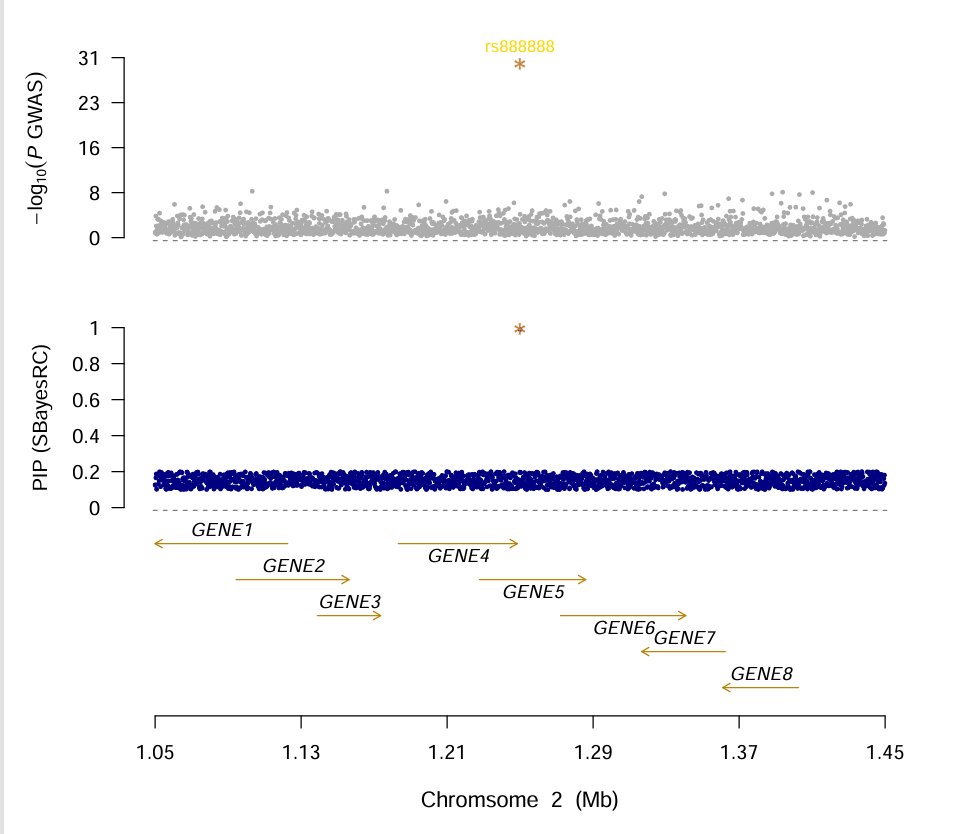
Locus plot for GWFM (genome wide fine mapping)
🧠 When you want draw locus plot of GWFM, follow scripts will meet your require
# ===========================================================================================//
# @Author : Loren Shi
# @Time : 2025/06/05 15:42:36
# @File : plot_GWFM.r
# @Mails : crazzy_rabbit@163.com
# @line : https://github.com/Crazzy-Rabbit
#
# R script to draw regional plot for genome wide fine mapping SNPs.
#
# Part of code are adopted from plot_smr script which provided by SMR
#
# Amendment:
# 2025/06/05
# 1. script completed
# 2. first realeased
# 3. fixed teh bug where the drawn gene was not in the middle of the genelayer when nrowgene=1
#
# Usages:
# source("plot_GWFM.r")
# PData <- ReadPvalueFromFiles(gwas="gwas_chrpos.gz", gwfm="gwaf.snpRes", glist="glist_hg19_refseq.txt", windowsize=200000, highlight="rs641221")
# pdf('gwfm_plot.pdf',width = 8,height = 8)
# MultiPvalueLocusPlot(data=PData)
# dev.off()
# ===========================================================================================//
is.installed <- function(mypkg){
is.element(mypkg, installed.packages()[,1])
}
# check if package "TeachingDemos" is installed
if (!is.installed("TeachingDemos")){
install.packages("TeachingDemos");
}
library(TeachingDemos)
library(data.table)
# parmeters for plot
genemove=0.01; txt=1.1; cex=1.3; lab=1.1; axis=1; top_cex=1.2;
GeneRowNum = function(GENELIST) {
BP_THRESH = 0.03; MAX_ROW = 5
# get the start and end position
GENELIST = GENELIST[!duplicated(GENELIST$GENE),]
START1 = as.numeric(GENELIST$GENESTART);
END1 = as.numeric(GENELIST$GENEEND)
STRLENGTH = nchar(as.character(GENELIST$GENE))
MIDPOINT = (START1 + END1)/2
START2 = MIDPOINT-STRLENGTH/250;
END2 = MIDPOINT+STRLENGTH/250
START = cbind(START1, START2);
END = cbind(END1, END2);
START = apply(START, 1, min);
END = apply(END, 1, max)
GENELIST = data.frame(GENELIST, START, END)
GENELIST = GENELIST[order(as.numeric(GENELIST$END)),]
START = as.numeric(GENELIST$START); END = as.numeric(GENELIST$END)
# get the row index for each gene
NBUF = dim(GENELIST)[1]
ROWINDX = rep(1, NBUF)
ROWEND = as.numeric(rep(0, MAX_ROW))
MOVEFLAG = as.numeric(rep(0, NBUF))
if(NBUF>1) {
for( k in 2 : NBUF ) {
ITERFLAG=FALSE
if(START[k] < END[k-1]) {
INDXBUF=ROWINDX[k-1]+1
} else INDXBUF = 1
if(INDXBUF>MAX_ROW) INDXBUF=1;
REPTIME=0
repeat{
if( ROWEND[INDXBUF] > START[k] ) {
ITERFLAG=FALSE
INDXBUF=INDXBUF+1
if(INDXBUF>MAX_ROW) INDXBUF = 1
} else {
ITERFLAG=TRUE
}
if(ITERFLAG) break;
REPTIME = REPTIME+1
if(REPTIME==MAX_ROW) break;
}
ROWINDX[k]=INDXBUF;
if( (abs(ROWEND[ROWINDX[k]]-START[k]) < BP_THRESH)
| ((ROWEND[ROWINDX[k]]-START[k])>0) ) {
MOVEFLAG[k] = 1
SNBUF = tail(which(ROWINDX[c(1:k)]==ROWINDX[k]), n=2)[1]
MOVEFLAG[SNBUF] = MOVEFLAG[SNBUF] - 1
}
if(ROWEND[ROWINDX[k]]<END[k]) {
ROWEND[ROWINDX[k]] = END[k] }
}
}
GENEROW = data.frame(as.character(GENELIST$GENE),
as.character(GENELIST$ORIENTATION),
as.numeric(GENELIST$GENESTART),
as.numeric(GENELIST$GENEEND),
ROWINDX, MOVEFLAG)
colnames(GENEROW) = c("GENE", "ORIENTATION", "START", "END", "ROW", "MOVEFLAG")
return(GENEROW)
}
ReadPvalueFromFiles <- function(gwas, gwfm, glist, windowsize=500000, highlight) {
gwas1 = fread(gwas)[, .(CHR, POS, SNP, p)]
colnames(gwas1) = c("CHR", "BP", "SNP", "p")
gwas1$CHR = as.character(gwas1$CHR)
gwas2 = fread(gwfm)[, .(Chrom, Position, Name, PIP)]
colnames(gwas2) = c("CHR", "BP", "SNP", "p")
gwas2$CHR = as.character(gwas2$CHR)
snp_gwas1 = gwas1[gwas1$SNP == highlight, ];
chrom = unique(snp_gwas1$CHR)
if (nrow(snp_gwas1) == 0) {stop("highlight SNP not found in file1, please check it!")}
BP1 = snp_gwas1$BP
start1 = BP1 - windowsize;
end1 = BP1 + windowsize;
file1 = gwas1[gwas1$CHR == chrom & gwas1$BP > start1 & gwas1$BP < end1, ]
snp_gwas2 = gwas2[gwas2$SNP == highlight, ];
if (nrow(snp_gwas2) == 0) {stop("highlight SNP not found in file2, please check it!")}
BP2 = snp_gwas2$BP
start2 = BP2 - windowsize;
end2 = BP2 + windowsize;
file2 = gwas2[gwas2$CHR == chrom & gwas2$BP > start2 & gwas2$BP < end2, ]
start = max(c(start1, start2), na.rm=T) / 1e6;
end = max(c(end1, end2), na.rm=T) / 1e6;
glist = fread(glist)
glist[,2] = glist[,2]/1e6;
glist[,3] = glist[,3]/1e6;
colnames(glist) = c("CHR", "GENESTART", "GENEEND", "GENE", "ORIENTATION")
glist = glist[glist$CHR == chrom & glist$GENESTART >= start & glist$GENEEND <= end, ]
return_lsit = list(file1=file1, file2=file2, SNP=highlight, glist=glist, CHR=chrom)
}
MultiPvalueLocusPlot <- function(data) {
gwas1 = data$file1;
gwas2 = data$file2;
pXY1 = -log10(gwas1$p);
yMAX = ceiling(max(pXY1, na.rm=T)) + 1;
pXY2 = gwas2$p;
yMAX2 = ceiling(max(pXY2, 1, na.rm=T));
glist = data$glist;
generow = GeneRowNum(glist);
num_row = max(as.numeric(generow$ROW));
offset_map = ceiling(yMAX);
offset_map = max(offset_map, num_row*2.5);
offset_pip = yMAX / 2.5;
dev_axis = 0.1*yMAX;
if (dev_axis < 1.5) dev_axie=1.5;
yaxis.min = -offset_map - offset_pip - dev_axis - yMAX;
yaxis.max = yMAX + ceiling(offset_pip) + 1;
gwasBP1 = as.numeric(gwas1$BP) / 1e6;
gwasBP2 = as.numeric(gwas2$BP) / 1e6;
xmin = min(c(gwasBP1, gwasBP2), na.rm=T) - 0.001;
xmax = max(c(gwasBP1, gwasBP2), na.rm=T) + 0.001;
xlab = paste("Chromsome ", data$CHR, " (Mb)")
#------------------- plot gwas layer ----//
ylab1 = expression(-log[10] (italic(P) * " GWAS"))
par(mar=c(5,5,3,2), xpd=TRUE);
plot(gwasBP1, pXY1, pch=20, xaxt="n", yaxt="n", bty="n", ylim=c(yaxis.min, yaxis.max),
xlim=c(xmin, xmax), xlab=xlab, ylab="", cex.lab=lab, cex.axis=axis,cex=0.6, col="gray68");
devbuf1 = yMAX/4;
xticks = round(seq(xmin, xmax, length.out=6), 2);
axis(1, at=xticks, lwd=1);
axis(2, at=seq(0, yMAX, by=devbuf1), labels=round(seq(0, yMAX, devbuf1), 0), las=1, lwd=1);
mtext(ylab1, side=2, line=3, at=(yMAX*1/2));
segments(x0=xmin, y0=-0.5, x1=xmax, y1=-0.5, col="grey50", lwd=1, lty=2);
snp1 = gwas1[SNP == data$SNP, ]
snpBP1 = snp1$BP / 1e6;
snpP1 = -log10(snp1$p);
points(snpBP1, snpP1, pch="*", col="peru", cex=2);
text(snpBP1, snpP1, labels=snp1$SNP, col="gold", pos=3, cex=0.8, offset=0.5);
#------------------- plot PIP layer ----//
ylab2 = "PIP (SBayesRC)"
axis.start = 0;
axis.start = axis.start - yMAX - offset_pip - dev_axis;
pXY2buf = pXY2 / yMAX2*yMAX + axis.start;
par(new=TRUE);
plot(gwasBP2, pXY2buf, pch=20, xaxt="n", yaxt="n", bty="n", ylim=c(yaxis.min, yaxis.max),
xlim=c(xmin, xmax), ylab="", xlab="", cex.lab=lab, cex.axis=axis, cex=0.6, col="navy");
devbuf2 = yMAX2/5;
axis(2, at=seq(axis.start, (axis.start+yMAX), yMAX/5), labels=seq(0, yMAX2, devbuf2), las=1, lwd=1);
mtext(ylab2, side=2, line=3, at=((axis.start + axis.start + yMAX)/2));
segments(x0=xmin, y0=axis.start-0.5, x1=xmax, y1=axis.start-0.5, col="grey50", lwd=1, lty=2);
snp2 = gwas2[SNP == data$SNP, ]
snpBP2 = snp2$BP / 1e6;
snpP2 = snp2$p / yMAX2*yMAX + axis.start;
points(snpBP2, snpP2, pch="*", col="peru", cex=2);
#------------------- plot gene layer ----//
num_gene = dim(generow)[1]
dist = offset_map / num_row;
for (k in 1:num_row) {
generowbuf = generow[which(as.numeric(generow[, 5]) == k), ]
xstart = as.numeric(generowbuf[, 3])
xend = as.numeric(generowbuf[, 4])
snbuf = which(xend - xstart < 1e-3)
if (length(snbuf) > 0) {
xstart[snbuf] = xstart[snbuf] - 0.0025
xend[snbuf] = xend[snbuf] + 0.0025
}
xcenter = (xstart+xend)/2
xcenter = spread.labs(xcenter, mindiff=0.01, maxiter=1000, min=xmin, max=xmax)
num_genebuf = dim(generowbuf)[1]
if (num_row == 1) {
for (l in 1:num_genebuf) {
ofs=0.3;
if (l%%2 == 0) ofs=-0.8;
ypos = offset_map/2 + yaxis.min - 1;
code = 1;
if (generowbuf[l,2] == "+") code=2;
arrows(x0=xstart[l], y0=ypos, x1=xend[l], y1=ypos, code=code, length=0.07, ylim=c(yaxis.min, yaxis.max),
col=colors()[75], lwd=1)
movebuf = as.numeric(generowbuf[l, 6]) * genemove
text(x=xcenter[l]+movebuf, y=ypos, label=substitute(italic(genename), list(genename=as.character(generowbuf[l,1]))), pos=3, offset=ofs, col="black", cex=0.9)
}
} else if (num_row > 1) {
for (l in 1:num_genebuf) {
ofs=0.3
if(l%%2==0) ofs=-0.8
m = num_row - k
ypos = m*dist + yaxis.min
code = 1;
if (generowbuf[l,2] == "+") code = 2;
arrows(x0=xstart[l], y0=ypos, x1=xend[l], y1=ypos, code=code, length=0.07, ylim=c(yaxis.min, yaxis.max),
col=colors()[75], lwd=1)
movebuf = as.numeric(generowbuf[l,6]) * genemove
text(x=xcenter[l]+movebuf, y=ypos, label=substitute(italic(genename), list(genename=as.character(generowbuf[l,1]))), pos=3, offset=ofs, col="black", cex=0.9)
}
}
}
}
Example data as follow:
set.seed(123)
n_snps <- 10000
n_genes <- 10
# SNP pos
positions <- as.integer(seq(500000, 2500000, length.out = n_snps))
snp_ids <- paste0("rs", 1000000 + 0:(n_snps - 1))
# highlight SNP(GWAS)
highlight_snp <- data.frame(
CHR = "2",
POS = 1250000,
SNP = "rs888888",
p = 1e-30
)
# GWAS summary
pvals <- pmax(rbeta(n_snps, 0.4, 5), 1e-8)
gwas_df <- data.frame(
CHR = rep("2", n_snps),
POS = positions,
SNP = snp_ids,
p = pvals
)
gwas_df <- rbind(gwas_df, highlight_snp)
write.table(gwas_df, file = "sim_gwas_chr2.gz", sep = "\t", row.names = FALSE, quote = FALSE)
# highlight SNP(PIP)
highlight_pip <- data.frame(
Chrom = "2",
Position = 1250000,
Name = "rs888888",
PIP = 0.99
)
# PIP data, finemapping result
pip_values <- round(runif(n_snps, 0.1, 0.2), 4)
gwfm_df <- data.frame(
Chrom = rep("2", n_snps),
Position = positions,
Name = snp_ids,
PIP = pip_values
)
gwfm_df <- rbind(gwfm_df, highlight_pip)
write.table(gwfm_df, file = "sim_gwfm.snpRes", sep = "\t", row.names = FALSE, quote = FALSE)
# gene list
gene_starts <- as.integer(seq(1050000, 1450000, length.out = n_genes))
gene_lengths <- sample(30000:80000, n_genes, replace = TRUE)
gene_ends <- gene_starts + gene_lengths
gene_df <- data.frame(
CHR = rep("2", n_genes),
GENESTART = gene_starts,
GENEEND = gene_ends,
GENE = paste0("GENE", 1:n_genes),
ORIENTATION = sample(c("+", "-"), n_genes, replace = TRUE)
)
write.table(gene_df, file = "sim_glist_chr2.txt", sep = "\t", row.names = FALSE, quote = FALSE)
# run script
source("plot_GWFM.r")
PData <- ReadPvalueFromFiles(gwas="sim_gwas_chr2.gz", gwfm="sim_gwfm.snpRes", glist="sim_glist_chr2.txt", windowsize=200000, highlight="rs888888")
pdf('gwfm_plot.pdf',width = 8,height = 8)
MultiPvalueLocusPlot(data=PData)
dev.off()
Job submit in supercompute
🧠 When your job number is very large, follow scripts will meet your require 🔁 An ldsc example for large number job submit (if job can split chromosome)
#! /bin/bash
ldsc="/public/home/shilulu/software/ldsc"
bfile="/public/share/wchirdzhq2022/Wulab_share/LDSC/1000G_EUR_Phase3_plink/1000G.EUR.QC"
ldsc_dir="/public/share/wchirdzhq2022/Wulab_share/LDSC"
files=( *.GeneSet )
total=${#files[@]}
batch_size=2
i=0
while [ $i -lt $total ]; do
batch=()
job_ids=()
for ((j=0; j<batch_size && (i + j) < total; j++)); do
file=("${files[$((i + j))]}")
annot=$(basename -- "${file}" ".GeneSet")
cmd1="python ${ldsc}/make_annot.py \
--gene-set-file ${annot}.GeneSet \
--gene-coord-file ${ldsc}/Gene_coord.txt \
--bimfile ${bfile}.{TASK_ID}.bim \
--windowsize 100000 \
--annot-file ${annot}.{TASK_ID}.annot.gz"
sub1=$(qsubshcom "$cmd1" 1 10G ldsc_anot 1:00:00 "-array=1-22")
cmd2="python ${ldsc}/ldsc.py --l2 \
--bfile ${bfile}.{TASK_ID} \
--print-snps ${ldsc_dir}/listHM3.txt \
--ld-wind-cm 1 \
--annot ${annot}.{TASK_ID}.annot.gz \
--thin-annot \
--out ${annot}.{TASK_ID}"
jobid=$(qsubshcom "$cmd2" 1 10G ldsc_comp 1:00:00 "-array=1-22 -wait=$sub1")
echo "🚀 Submitted job $jobid for $annot, waiting for it finish..."
job_ids+=("$jobid")
done
echo "⏳ Waiting for jobs: ${job_ids[*]}"
times=0
while true; do
sleep 30
all_done=true
for jid in "${job_ids[@]}"; do
if squeue -j "$jid" | grep -q "$jid"; then
all_done=false
times=$((times + 30))
echo "⏳ Jobs in batch still running ${times}s..."
break
fi
done
if $all_done; then
echo "✅ All jobs in batch completed."
break
else
echo "⏳ Still waiting for some jobs in batch to finish..."
fi
done
i=$((i + batch_size))
done
🔁 An t-test example for large number job submit
#! /bin/bash
THRESHOLD=60
OUTPUT_FILE_THRESHOLD=2400
ARRAY_SIZE=2
JOB_ID_FILE="job_id.txt"
MAX_JOB_ID=100
OUT_DIR="t_stat_blocks_cell"
if [ ! -f "$JOB_ID_FILE" ]; then
echo 1 > "$JOB_ID_FILE"
fi
cell_type_count=$(wc -l < cell_type.txt)
submit_per_loop=$((ARRAY_SIZE * cell_type_count))
while true; do
output_file_count=$(ls $OUT_DIR 2>/dev/null | wc -l)
current_jobs=$(squeue | grep -v "JOBID" | wc -l)
if [ "$output_file_count" -ge "$OUTPUT_FILE_THRESHOLD" ]; then
echo "✅ Output file count ($output_file_count) ≥ threshold ($OUTPUT_FILE_THRESHOLD), exit."
exit 0
fi
start_id=$(cat "$JOB_ID_FILE")
end_id=$((start_id + ARRAY_SIZE -1))
if [ "$start_id" -gt "$MAX_JOB_ID" ]; then
echo "🚫 All jobs submitted (up to $MAX_JOB_ID), waiting for output files..."
sleep 600
continue
fi
if [ "$current_jobs" -lt "$THRESHOLD" ] && [ $((current_jobs + submit_per_loop)) -le "$THRESHOLD" ]; then
echo "🚀 Submitting jobs: ID range $start_id to $end_id"
echo "Current jobs: $current_jobs, Will submit: $submit_per_loop"
while read -r idx cell; do
qsubshcom "Rscript t-stat.r {TASK_ID} $idx" 10 100G t-stat 10:00:00 "-array=${start_id}-${end_id}"
done < cell_type.txt
echo $((end_id + 1)) > "$JOB_ID_FILE"
sleep $((60 + RANDOM % 30))
else
echo "⏳ Current job number too high or not enough capacity, wait and retry"
sleep 60
fi
done
Stereo file to scanpy
🧠 When your want transfer stereo-seq file gef to scanpy or seurat, follow scripts will meet your require
conda activate st
# conda create --name st python=3.8
# conda install stereopy -c stereopy -c grst -c numba -c conda-forge -c bioconda
import stereo as st
import scanpy as sc
import warnings
warnings.filterwarnings('ignore')
# read the GEF file
data_path = 'D02567D4.gef'
data = st.io.read_gef(file_path=data_path, bin_size=50)
data.tl.cal_qc()
data.tl.raw_checkpoint()
data.tl.sctransform(res_key='sctransform', inplace=True)
# clustering
data.tl.pca(use_highly_genes=False, n_pcs=30, res_key='pca')
data.tl.neighbors(pca_res_key='pca', n_pcs=30, res_key='neighbors')
data.tl.umap(pca_res_key='pca', neighbors_res_key='neighbors', res_key='umap')
data.tl.leiden(neighbors_res_key='neighbors', res_key='leiden')
# write as stereopy h5ad
st.io.write_h5ad(
data,
use_raw=True,
use_result=True,
key_record=None,
output='./D02567D4.h5ad')
data_path = './D02567D4.h5ad'
data = st.io.read_h5ad(
file_path=data_path,
flavor='stereopy',
use_raw=True,
use_result=True)
#----------------
# transfer to h5ad of scanpy
adata = st.io.stereo_to_anndata(data, flavor='scanpy',output='out.h5ad')
adata.layers["count"] = adata.X.copy()
adata.obs["annotation"] = 'teeth'
adata.write("D02567D4_scanpy.h5ad")
liftover in R
#---- liftover hg38 to hg19
library(rtracklayer)
library(GenomicRanges)
regions = unique(as.character(merged_dt$Gene))
regions_str = strsplit(regions, "-")
bed_df = do.call(rbind, lapply(seq_along(regions), function(i) {
x = regions_str[[i]]
chr = x[1]
start = as.numeric(x[2]) - 1
end = as.numeric(x[3])
peak_id = regions[i]
return(data.frame(chr=chr, start=start, end=end, peak_id=peak_id))
}))
gr <- GRanges(seqnames = bed_df$chr,
ranges = IRanges(start=bed_df$start, end=bed_df$end),
peak_id = bed_df$peak_id)
chain <- import.chain("~Wulab/refGenome/hg38/hg38ToHg19.over.chain")
gr_new_list <- liftOver(gr, chain)
gr_new <- unlist(gr_new_list)
gr_clean <- gr_new[elementNROWS(gr_new_list) == 1]
df_clean <- as.data.frame(gr_clean)[, c("seqnames", "start", "end", "peak_id")]
df_clean_unique <- df_clean[!duplicated(df_clean$peak_id), ]
# peak coord file
coords <- df_clean_unique[, c("peak_id", "seqnames", "start", "end")]
colnames(coords) <- c("GENE", "CHR", "START", "END")
coords$CHR <- sub("^chr", "", coords$CHR)
fwrite(coords, "Peak_coord_hg19.txt", sep="\t")
QC for GWAS summary
sums_col_treate <- function(gwas, SNP, A1, A2, freq, P,
CHR = NULL, POS = NULL,
BETA = NULL, SE = NULL,
OR = NULL, Z = NULL, N=NULL) {
gwas_col = c(
CHR = CHR,
POS = POS,
SNP = SNP,
A1 = A1,
A2 = A2,
freq = freq,
b = BETA,
se = SE,
OR = OR,
Z = Z,
p = P,
N = N)
gwas_col = gwas_col[!sapply(gwas_col, is.null)]
missing_required = setdiff(c("SNP", "A1", "A2", "freq", "p"), names(gwas_col))
if (length(missing_required) > 0) {
stop("Missing required columns: ", paste(missing_required, collapse = ", "))
}
has_b_se = all(c("b", "se") %in% names(gwas_col))
has_or = "OR" %in% names(gwas_col)
has_z = "Z" %in% names(gwas_col)
if (!(has_b_se || has_or || has_z)) {
stop("Must provide at least one of the following: (BETA + SE), OR, or Z.")
}
gwas = gwas[, unname(gwas_col), with=FALSE]
setnames(gwas, old = unname(gwas_col), new = names(gwas_col))
valid_p <- !is.na(gwas$p) & gwas$p >= 0 & gwas$p <= 1
if (has_b_se){
gwas = gwas[valid_p & !is.na(SNP) & !is.na(b) & !is.na(se) & b != 0, ]
} else if (has_or){
gwas = gwas[valid_p & !is.na(SNP) & !is.na(OR), ]
} else if (has_z){
gwas = gwas[valid_p & !is.na(SNP) & !is.na(Z), ]
}
return(gwas)
}
🧠 How to use it
library(data.table)
gwas = fread("Ever_smoke.fastGWA.gz")
clean = sums_col_treate(gwas, "variant_id", "effect_allele", "other_allele", "effect_allele_frequency", "p_value", BETA="beta", SE="standard_error" N="N")
clean = sums_col_treate(gwas, "SNP", "A1", "A2", "AF1", "P", BETA="BETA", SE="SE", N="N")
clean = clean[, .(SNP, A1, A2, freq, b, se, p, N)]
fwrite(clean, "../cleanGWAS/clean_ever_smoke.fastGWA.gz", sep="\t")